 Início
Início
 macenadoctor
macenadoctor- Mensagens : 129
Data de inscrição : 27/05/2021
Tipos de anemia
Sáb Jul 31, 2021 10:52 am
- Conceitos iniciais:
- Carencial:
- Hematopoiese medular: célula formada nos ossos
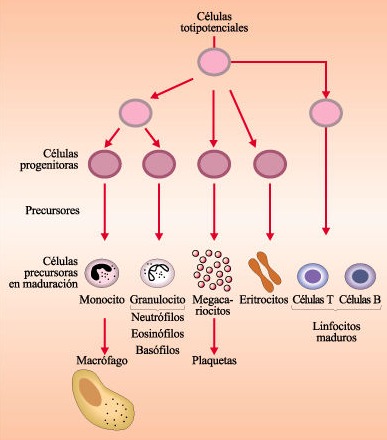
Combustíveis importantes para eritropoiese:
Eritropoietina (estímulo)
Ferro (hemoglobina- Hb)
Ácido fólico/ vitamina B12 (síntese de DNA)
Reticulócito é lançado na corrente e depois se desenvolve em hemácias, por isso, é medido para saber o paciente está responsivo aos medicamentos.
A hemácia é normalmente destruída em 120 dias pelo baço (hemocaterese- morte natural) ou sofre hemólise (morte prematura)
- Hemolítica:
- Hemocatarese: fisiológico (> 120 d)
Hemólise: patológico (< 120 d)
Passou devagar pelo baço, macrófago mata
Fatores compensatórios da hemólise:
1) Hiperplasia da medula
2) estoques de Fe, B12 e folato limitantes por não serem bem aproveitados
3) O corpo não consegue compensar se as hemácias morrerem com menos de 20 dias
↓ Hemácias devido à hemólise -> Hipóxia renal -> ↑ Epo -> MO tem hiperplasia em 6-8x -> ↑ reticulócitos; ↑ VCM -> ↑ hemólise
Causas de reticulocitose:1) anemias hemolíticas
2) sangramento agudo
3) melhora da ferropriva
Pancitopenia:
Anemia aplásica ou leucemia aguda
Pancitopenia leve:
Megaloblástica, mielodisplasia, neoplasia hematológica ou não hematológica metastática
Plaquetopenia:
Anemia autoimune, com evolução à síndrome de Evans
Leucoeritroblastose:
Ocupação medular por processo patológico (mielofibrose idiopática, neoplasia metastática ou mesmo uma infecção disseminada)
- Passos no diagnóstico da anemia:
- Primeiro passo:
- Reticulócitos: se houver ou não combustíveis
Se há reticulócitos (> 2%), é uma anemia hiperproliferativa, se não os há (≤ 2%), é uma hipoproliferativa- Hiperproliferativa:
- Anemia hemolítica
Sangramento agudo
- Hipoproliferativa:
- DRC (baixa eritropoietina)
Anemias carenaisis (ferropriva, B12, folato)
Anemia de doença crônica
Anemia sideroblástica
- Segundo passo:
- Morfologia da hemácia (VCM, HCM)
Basicamente irão nos fazer sobre as condições da Hb e Fe, o VCM é sobre o tamanho da hemácia, e a HCM é sobre a coração da célula.
A Hb é formada por um grupo heme ligado a ferro + protoporfirina
+ Globina formada por 2 cadeias alfa e 2 beta
Se estiver em baixa de ferro podemos estar diante de uma anemia ferropriva ou por doença crônica
Se na protoporfirina, pode ser uma sideroblástica
Quando é nas cadeias da globina, é uma talassemia.
- Leitura do hemograma:
Série vermelha Hemácias 4-6 milhões/mm³ Anemia Hb 12-17 g/dL Ht 36-50% Reticulócitos 0,5-2% Hipo/hiper VCM 80-100 f/L Micro/macro HCM 28-32 pg Hipo/hiper CHCM 32-35 g/dL Similar à HCM RDW 10-14 % Índice de anisocitose Série branca Leucócitos 5000-11000/mm³ Plaquetas 150k-400k/mm³
- Anemia hipoproliferativa:
- Anemia ferropriva:
- Clínica:
- Síndrome anêmica: palidez, astenia, cefaleia, angina ...
Carência nutricional: glossite, queilite angular
Perversão do apetite
Coiloníquia (unha em colher)
Disfagia (Plummer Vinson)
Sangramento crônico (TGI) em adultos
Coronopatas-> angina pectoris
Cardiopatas-> insuficiência cardíaca
Pneumopatas-> dispneia
Cerebrovasculopatas-> rebaixamento de consciência
Ainda há a possibilidade da má absorção de ferro, como ocorre na anastomose Billroth II que impede uma absorção importante do ferro. Lembrando que desse, temos a doença celíaca que impede a absorção.
Ancilostomídeos: Necatur americanus e Ancylostoma duodenale (mais importantes no Brasil) porque se aderem à mucosa duodenal. O Trichuris trichiura (tricocéfalo) pode causar múltiplas lesões hemorrágicas no cólon.
- Laboratório:
- Cinética do ferro:
- 1) ↓ ferritina < 30 (VR: 30-100 ng/mL)- devido ao esgotamento de estoque do ferro
2) ↑ transferrina e ↑ TIBC > 360 (VR: 250-360 mcg/dL)
3) ↓ ferro sérico < 30 (VR: 60-150 mcg/dL)
↓ Sat de transferrina < 10% (VR: 20-40%)
- Hemograma:
- 4) Anemia- normo/normo de início e evolui para micro/hipo (apresentação clássica)
5) ↑ RDW > 14 (VR: 10-14%) - anisocitose
6) ↑ Plaquetas (trombocitose)
- Tratamento:
- Investigar causa a tratar
Crianças: prematuridade (porque no fim da gestação é que o bebê absorver muito ferro), desmame (há ferro de boa absorção no leite materno), ancilostomíase
Adultos: gravidez, hipermenorreia, má absorção (doença celíaca), perda crônica de sangue pelo TGI
Repor ferro (sulfato ferroso: 20% Fe elementar)
Dose: 120-200 mg/d de Fe elementar
* Criança: 3-5 mg/kg/d Fe elementar
Resposta: reticulócitos (pico 7-10d)
Normaliza: ± 2 meses
Duração: por 6 meses OU até a ferritina > 15-50 ng/mL
Ferro parenteral (refratários ou má absorção)
Gluconato férrico de sódio e ferro-sucrose
Antes usava-se o ferro dextrano, mas há alta chance de anafilaxia
- Talassemia:
- Definição:
- Defeito na quantidade de globinas
4 hemes + 4 globinas -> Hb
ααββ = HbA 97%
ααδδ = HbA2 2%
ααγγ = HbF 1% - fetal, prevalece até os 6 meses de vida
- Betatalassemia:
- βº e β +
βº não produz nada
β + produz um β fraco
Por haver essa falha nos β, então a HbA não será formada, ou será mal formada, levando ao acúmulo de α que é tóxico;
Eritropoiese ineficaz (hemólise na MO)
Hemólise (baço e fígado)
HCM ↓; VCM ↓; RDW normal
Acúmulo de Ferro
Diagnóstico: Eletroforese de Hb com alta de HbF e de HbA2- Clínica:
- Betatalassemia major (βº βº; βº β +):
Cooley
HbF em 95%
Anemia hemolítica grave após 6 meses
Hepatoesplenomegalia/baixa estatura
Expansão da MO (deformidades)- fasceis de esquilo
Há muita morte de hemácia e a MO doente não capta o Ferro livre, gerando a hemocromatose
Tratamento: Ácido fólico/ quelante FC, esplenectomia ou transplante de MO
Betatalassemia intermedia (β + β +):
Tratamento: ácido fólico e quelante do Ferro
Betatalassemia minor (β βº; β β +)- traço betatalassêmico:
Só anemia hipo/micro
Tratamento é acompanhamento
PARA DIFERENCIAR A TALASSEMIA DA FERROPRIVA É SÓ ANALISAR O RDW, PORQUE NA FERROPRIVA ELE ESTÁ ALTO, ENQUANTO NA MINOR ESTÁ NORMAL HAJA VISTA QUE AS CÉLULAS NESSE CASO SÃO SEMPRE IGUAIS.
- Alfatalassêmica:
- Deleção ≥ 1 α (ao todo são 4 α)
Hemácia pequena, sobra β e γ (tóxicos)
Clínica e tratamento semelhantes a da betatalassemia
Sintomas se ≥ 3 deleções (desde nascido)- Clínica:
- Assintomático (α α α -)
α talassemia minor (α α - - )
Micro/hipo
Eletroforese de Hb normal
α talassemia intermédia (α - - -)
Doença de HbH (α β β β)-
Anemia hemolítica moderada/grave
Eletroforese: Hb 5-40% HbH
Hidropsia fetal (- - - -)
Incompatível com a vida
Eletroforese Hb: 80% HbBart
- Anemia megaloblástica:
- Definição:
- Déficit de ácido fólico e/ou vitamina B12
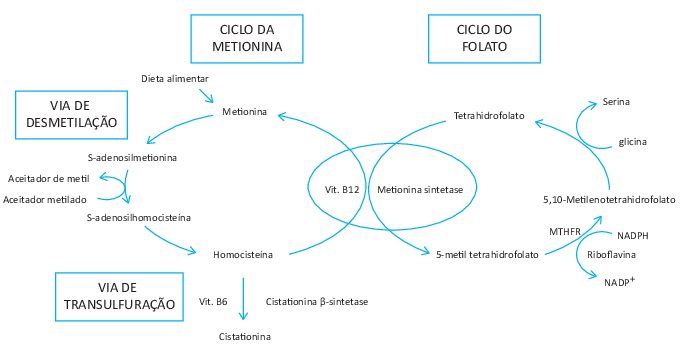
A deficiência impede a divisão celular, por isso são células grandes
Megaloblastos (células que não dividiram) -> eritropoiese ineficaz (hemólise na MO)
Macrocitose, pancitopenia (porque tem meia-vida curta), neutrófilos hipersegmentados (define diagnóstico)
Metabolismo do folato (presente em folhas)
MTHF- metil tetra hidrofolato
Deficiência -> má nutrição: alcoólotra, criança
↓ Absorção -> doença celíaca, fenitoína
↓ Regeneração -> metotrexato
↑ Necessidade -> gestante, hemólise crônica
Metabolismo de cobalamina (B12)
Carne (proteína/B12) -estômago-> Proteína + B12/Ligante R (saliva) -duodeno e pâncreas-> Ligante R + B12/Fator intrínseco (FI) vindo do estômago -íleo terminal-> B12/FI -> absorção
- Clínica:
- Causas: anemia perniciosa; vegetarianismo; gastrectomia; pancreatite crônica; doença ileal; Diphyllobothrium latum (presente no peixe cru)
Clínica: síndrome anêmica; glossite + quielite + diarreia
Deficiência de B12
Síndrome neurológica: neuropatia, mielopatia, demência;
Perda de sensibilidade vibratória
Degeneração cortical da medula: lesão corticoespinal + cordões posteriores
Ácido metilmalônico -B12-> Succinil-CoA (presente na mielinização)
A deficiência de B12 causa excesso de Ac. metilmalônico causa toxicidade neuronal
- Diagnóstico:
- Laboratório:VCM > 110 (muito provável)/ neutrófilos hipersegmentados
Normocrômica e ↑ RDW
↓ Plaquetas e ↓ leucócitos (pancitopenia leve)
↑ LDH e BI (eritropoiese ineficaz)
↑ hemocisteína: B12 e folato
↑ Ac. Metilmalônico: B12
Pleocariócitos = neutrófilos hipersegmentados
- Tratamento:
- Deficiência de B12: Repor B12 IM
Deficiência de Folato: Repor folato VO
Essas reposições causam hipocalemia
Dosamos reticulócitos 2 dias após porque é quando começa a surtir efeito significativo
- Anemia de doença crônica (inflamação):
- Epidemiologia:
- 2ª causa de anemia e 1ª em hospitalização
Inflamatória: 33-60%
Infecções 18-95%
Malignidades 30-63%
Associada com envelhecimento
Obesidade
DM
IC
- Fisiopatologia:
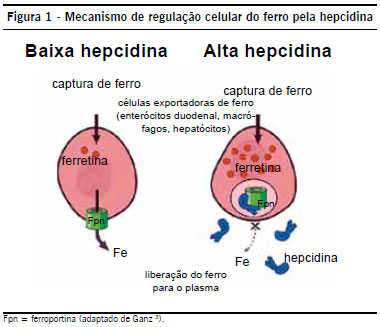
Macrófagos são importantes por reciclar Fe-> 95% do Fe diário
- Clínica:
- Astenia, dispneia, tonturas, anorexia, cefaleia
Palidez cutâneo-mucosa
Diversos e desafiador - múltiplos sintomas de etiologia base
- Exames complementares:
- Hemograma: anemia normo-normo 10-12 g/dL
Reticulócitos: baixos
Ferro sérico: baixo -> porque a hepcidina prende ferro
Transferrina: normal ou baixa (<300 mcg/dL)
Sat. transferrina: baixa (<20%)
Ferritina: normal ou elevada (>100 mcg/dL)- sua alta se dá pelo bloqueio da hepcidina
PCR: elevada (> 5 mg/dL)
Mielograma (ferro rosa; azul macrófago): excesso de ferro em macrófago é pela hepcidina
A hepcidina será liberada pelo fígado principalmente pela estimulação da IL-6 que é tão presente em doenças inflamatórias crônicas.
- Tratamento:
- Tratar a causa base: inflamatórias e autoimune
Infecciosas
Neoplasias
Obesidade
DM
Reposição de ferro: até ferritina < 100 mcg/dL e IST < 20%
Oral se função renal adequada
Venosa se má função renal
Evitar ferro em infecção ativa porque Fe ajuda o crescimento de macrófagos
Alfaeritropoetina:
Basicamente Doença renal crônica
Oncológico- em casos excepcionais
Cirurgia
Transfusão:Anemias graves, sintomáticas e risco de óbito
Avalia se há tempo para outro tratamento
Não guiar só por Hemoglobina e hematócrito
Tratar a causa base;
- Anemia sideroblástica:
- Definição:
- Causada por deficiência de protoporfirina
Hereditária
Adquirida (álcool, ↓ B6, ↑ cobre)
- Laboratório:
- ↑ Ferro sérico-> risco de hemocromatose
↑ Ferritina
↑ Sat. transferrina
Acúmulo de ferro na linhagem eritrocitária
Aspirado de MO (mielograma)-
Sangue periférico (hematoscopia)- corpúsculo de Pappenheimer
Na eritropoiese ocorre a hemoglobinização do citoplasma
2 Succinil CoA + 2 glicina ---- ALA sintase + Vit B6----> ALA -> Protoporfirina IV + Fe²+ -> Heme
O Fe²+ vem do Fe³+
Na anemia sideroblástica hereditária ocorre a deficiência de ALA sintase
Deficiência na conversão do ferro: Anemia sideroblástica adquirida idiopática
Essas duas causas fazem com que sobre muito Fe levando à formação do anel de ferro
Deficiência na B6: adquirida por etanol
Deficiência no cobre: impedindo a conversão do ferro- Microcítica:
- Hereditária
Deficiência de cobre
- Macrocítica:
- Idiopática
Álcool
- Tratamento:
- Heriditária: B6
Adquirida: Epo recombinante
Flebotomia
Quelantes do Fe
Adquirida: suspender agente agressor
- Anemia hiperproliferativa:
- Conceito geral:
- Clínica: Síndrome anêmica, esplonomegalia, icterícia leve, cálculo de bilirrubimato de cálcio (cálculo na bile)
Laboratório: Anemia normo; ↑ VCM; Reticulocitose; ↑ LDH; ↑ BI (devido à protoporfirina); ↓ Haptoglobina
Complicações: crises anêmicas agudas "de repente surge uma anemia +(...)"- Anemia + reticulocitopenia:
- 1) Aplásica-> causa: infecção (parvovírus B19)-> sem tratamento
2) Megaloblástica-> Causa e tratamento: ácido fólico; diagnóstico diferencial pela presença de neutrófilos hipersegmentados
- Anemia + injúria renal aguda:
- 3) Hemólise aguda grave-> Causas: ↓ G6PD; Loxocelles Necrose tubular aguda (NTA) com IRA oligúria "hemoglobinúria maciça com lesão renal"
- Anemia + esplenomegalia:
- 4) Sequestro esplênico: causa mais comum: anemia falciforme
- Coombs direto:
- Positivo (+)- autoimune:
- Além do Coombs direto, temos um microesferócito;
Anticorpos quentes (IgG) 75% Crioglutininas (IgM) Idiopática: maioria Micoplasma/Mononucleose/Macroglobulinemia Secundária: viral, LES, LLC Tratar a causa Drogas: penicilina,metildopa Plasmaferese Tratamento: causa, corticoide, Rituximab/esplenectomia Rituximab
- Negativo (-)- não autoimune:
Hereditárias Adquiridas Membrana: esferocitose Hbnúria paroxística noturna Enzima: ↓ G6PD Hiperesplenismo Hemoglobinopatias: falciforme e talassemias síndrome hemolítica urêmica (SHU) e a púrpura trombocitopênica trombótica (PTT)
- Esferocitose hereditária:
- Clínica: criança + esplenomegalia
Anemia hemolítica hipercrômica + esferócitos (↑ CHCM)
Diagnóstico: teste de fragilidade osmótica
Tratamento: esplenectomia parcial/total após 5-6 semanas se: anemia grave e/ou repercussão clínica
Antes da esplenectomia, ter certeza da vacinação da criança
- Deficiência de G6PD:
- Clínica: mais comum em homens
Anemia hemolítica após superoxidação
A G6PD atua ativando o glutation que guarda as células mortas, mas em caso de infecção as células morrem antes dos 120 dias, levando a essa superoxidação
Causas: infecção (+ comum), drogas (sulfa, primaquina, dapsona, naftalina e nitrofurantoína)
Diagnóstico: Corpúsculo de HEINZ
Medida da atividade de G6PD
Tratamento: prevenir falando o que causa essa superoxidação
- Hemoglobinúria paroxística noturna:
- Desordem genética adquirida extraútero
Fisiopatologia: mutação deixa hemácia + fácil de ser lisada por complemento
Clínica: homens de 30-40 a.
Tríade: hemólise, pancitopenia, tromboses abdominais
Diagnóstico: citometria de fluxo: ↓ CD55 CD59
Tratamento: Eculizumab (capaz de inibir complemento)
- Hemoglobinopatias:
- Anemia falciforme:
- Definição:
- Quando a hemácia perde O2 ou H2O, as HbS se enfileram numa só região da célula, causando a forma de foice;
Após 15 dias de vida da célula, esse formato é irreversível.
Hemácia afoiçada é um drepanócito que libera fator de agregação (aumenta a chance de oclusão) e andam em desordem causando lesão endotelial.
Cromossomo 16: α α
Cromossomo 11: β δ γ
Na falciforme, a β passa para a β S que faz parte da HbS
Quando se é heterozigoto faz-se HbS e HbA (traço falcêmico- sem sintomas, nem mesmo no hemograma)- Clínica:
- Começa após 6 meses de vida, porque até isso são HbF
- Crises vaso-oclusivas agudas:
- 1) Síndrome mão-pé: primeira manifestação falciforme
Idade: até 2-3 anos
Local: extremidades
Clínica: dor com dactilite
Raio X: normal
2) Crise óssea: crise álgica/osteoarticular + comum
Idade: > 3 anos
Local: ossos longos
Clínica: dor sem dactilite
Raio x: normal
3) Sequestro esplânico (esplenomegalia + anemia) (<5 anos)
Esplenomegalia: congestão esplênica (vascular)
Anemia: sequestro e hemólise esplênicas
O sequestro surge antes dos 5 anos, ocorre esplenomegalia com fibrose progressiva do baço
Atenção: Anemia falciforme em pacientes > 5 a. em 5% é por alta produção de HbF
4) Síndrome torácida aguda
Clínica: infiltrado pulmonar novo (parece uma pnemonia com evolução rápida) e um dos seguintes: febre, tosse, dor torácica, expectoração purulenta dispneia
SE PACIENTE TEM ANEMIA FALCIFORME E FICA COM FEBRE, INTERNA
5) AVE
Crianças: isquêmico
Adultos: hemorrágico
- Disfunções crônicas:
- 1) Lesão óssea: necrose avascular do osso (osteonecrose)
Osteomielite por Salmonella/S. aureus
Artrite séptica: normalmente pneumococo
2) Lesão renal: necrose de papila
GEFS
Ca medular
Hidratar: não hiper hidratar
O2: só se SatO2 < 92%
Analgesia: inclui opioides
ATB: β lactâmico se paciente tem febre, ↓ PA, Hb < 5, leuco > 30k
Tratamento crônico
Prevenção: ácido fólico, vacinação, penicilina VO até 5 anos ( começa em 2-3 meses)
Hidroxiureia: ↑ HbF, crises álgicas (> 2-3/a.), prevenção secundárias a Síndrome torácia aguda/AVE e anemia grave sintomática
Transplante de Medula Óssea: < 16 anos
Tratamento transfusional
Transfusão simples: anemia sintomática ou Hb <5,5 (criança), < 6 (adulto)
Exsanguíneo-transfusão
Aguda: quadros graves (AVE, STA)
Crônica (regulares) com alvo: Hb 9-10 e HbS < 30%
Fazer regulares se não respondeu a hidroxiureia e prevenção secundária a AVE/STA
- TGI:
- Pacientes > 50 a. com anemia ferropênica micro/hipo fazemos uma colonoscopia para encontrar a causa de um possível sangramento no TGI
A gastrite autoimune é a principal não causada pela H. Pylori, poupa o antro, as células parietais produtores de Fator Intrínseco são destruídas, atrapalhando a absorção de B12- causa anemia
- Anemia fisiológica:
- Características do RN:
- Policitêmico
Eritroblastêmico
Hipervolêmico
Hipersiderêmico
- RN no pós parto:
- ↑ PaO2 -> ↓ eritropoietina
↓Meia vida das hemácias
Esses dois pontos causam a anemia fisiológica
- Anemia fisiológica no RN a termo:
- Início em 7 dias
Nadir: 6-12 semanas
Normo/normo -> só mantém amamentação
Hb 9,5-11 g/dL
- Anemia fisiológica no RN pré-termo:
- Baixa reserva de ferro
Clampeamento precoce
Espoliação do sangue
- Anemia na prematuridade:
- Nadir: 3-6 semanas
Hb: 7,9 g/dL
Concentrado de hemácias no SN como tratamento
- Metabolismo do ferro:
O Ferro heme é absorvido diretamente no duodeno
O ferro não heme é convertido por facilitadores como a vitamina C, ou pode ser inibido por fitatos (presente em cereais e leguminosas), taminos (vinho), cálcio e fósforo (presentes no leite, soja e ovo)Armazenagem Fonte Ferro heme (Fe2+) Ferro não heme (Fe3+) Hb Alimentação Carnes Vegetais Mioglobina Reciclagem Vísceras Ferritina Transferrina
- Fatores de risco:
- Prematuridade
AME < 6 meses
Abaixamento do nível socioeconômico
Alimentação pobre em ferro
Falta da profilaxia do ferro
Adolescência
A grande causa de anemia ferropriva é a alta ingestão do leite de vaca
- Clínica:
- Deficiência de ferro: astenia, dor MMII, unhas rugosas, estomatite angular, perversão do apetite, prejuízo cognitivo, distúrbio de conduta
Anemia: apatia, irritabilidade, palidez, sopro, taquicardia, intolerência a exercícios
- Diagnóstico:
- Exames laboratoriais
- Tratamento:
- 3-5 mg/kg/d de ferro antes das refeições
Tempo mínimo de 8 semanas
Objetivo: ferritina > 15 ug/dL
Monitorização
Hemograma e reticulócitos sendo reavaliados a cada 30-60 dias
Perfil do ferro e ferritina sendo reavaliados a cada 30-90 dias
Reticulócitos tem seu pico após 7 dias
Hb- 30 dias
Ferritina: 2-6 meses
Efeitos adversos: gosto metálico, náuseas /vômitos, pirose/dispepsia, plenitude abdominal, diarreia/obstipação
opções: sair férricos e aminoquelatos
- Profilaxia:
- AME até 6 meses
Alimentação com ferro
Segundo MS
RN a termo com peso adequado: inicia com 6 meses; 1 mg/kg/d de ferro elementar e mantém até 24 meses
Segundo SBP (2018): inicia com 3 meses, indepente do AME, 1 mg/kg/d ferro elementar e mantém até 24 meses
Ressalva para que aqueles que ingerem mais de 500 mL/d de fórmula não precisam dessa profilaxia
RN prematuros/baixo peso
No 1º ano: inicia em 15 d-2m de vida
No 2º ano: 1 mg/kg/dBaixo peso 2 mg/kg/d Muito baixo peso 3 mg/kg/d Extremo baixo peso 4 mg/kg/d
- macenadoctor
- Mensagens : 129
Data de inscrição : 27/05/2021
Re: Tipos de anemia
Ter Ago 03, 2021 2:46 pm
Existem dois tipos de hemocromatose:
A primária (pode ser subdividida em 4 outros grupos, mas eles se referem simplesmente a qual enzima está defeituosa) é uma forma hereditária. O que acontece é que a hepcidina acaba não tendo sua atividade bem feita, fazendo que com que todo ferro absorvido esteja em ação.
Diagnóstico: Ferritina sérica, Ferro sérico em jejum e saturação da transferrina
Exame genético
Biópsia hepática
Tratamento: Flebotomia que retira sangue semanalmente do paciente, em torno de 500mL
A sobrevida pode até ser prolongada, mas não evita o carcinoma hepatocelular;
Existe a hemocromatose adquirida, muito comum quando o paciente passa por muitas transfusões sanguíneas.
A primária (pode ser subdividida em 4 outros grupos, mas eles se referem simplesmente a qual enzima está defeituosa) é uma forma hereditária. O que acontece é que a hepcidina acaba não tendo sua atividade bem feita, fazendo que com que todo ferro absorvido esteja em ação.
Diagnóstico: Ferritina sérica, Ferro sérico em jejum e saturação da transferrina
Exame genético
Biópsia hepática
Tratamento: Flebotomia que retira sangue semanalmente do paciente, em torno de 500mL
A sobrevida pode até ser prolongada, mas não evita o carcinoma hepatocelular;
Existe a hemocromatose adquirida, muito comum quando o paciente passa por muitas transfusões sanguíneas.
- macenadoctor
- Mensagens : 129
Data de inscrição : 27/05/2021
Anemia hemolítica autoimune (AHAI)
Ter Ago 03, 2021 10:40 pm
- Definição:
- Autoanticorpos reagem contra grupos sanguíneos
As IgG dos eritrócitos ligam-se a receptores FcγRI de macrófagos que opsonizam as hemácias.
Isso também pode ocorrer pelo complemento C3b
- Etiologia:
- Ac Quente- IgG:
- Esses anticorpos se ligam com o corpo em temperatura de 37ºC
Pode causar hemólise extravascular
Metade dos casos é idiopático.
Pode surgir por drogas (alfametildopa, fludarabina), LES, leucemia linfocítica crônica, linfoma não Hodgkin etc.- Clínica:
- 50-60 a.
Mulheres (estrogênio piora)
Icterícia, quadro muito variante de assintomático até episódio hemolítico agudo
Síndrome de Evans:Púrpura trombocitopênica autoimune + Anemia autoimune (AHAI)
Laboratório: reticulocitose; VCM baixo ou normal
Microesferócitos no esfregaço sanguíneo- se deve à fagocitose parcial do baço, retirando pedaços de membrana eritrocitária
- Diagnóstico:
- Coombs direto- uma gota de sangue do paciente, incubando-se a 37°C. Se for detectada aglutinação macroscópica, o teste é considerado positivo
- Tratamento:
- Específico: Glicocorticoides (prednisona) 1-2 mg/kg/d ou 40 mg/m²
Efeito dos corticoides:
(1) reduzem a afinidade dos receptores FcγRI dos macrófagos esplênicos – sendo este o responsável pelo início da resposta nos primeiros quatro dias; (2) reduzem a afinidade dos anticorpos IgG pelos antígenos da membrana eritrocítica; (3) diminuem a produção de anticorpos IgG (este é o efeito responsável pela resposta mais tardia, porém, mais duradoura).
Se refratário, usamos rituximabe (ac monoclonal) ou esplenectomia. Se mesmo após isso continuarem com anemia hemolítica é por causa da opsonização pelas células de Kupffer hepáticas.
Em hemotransfusão, o sangue do paciente aglutina com todas as amostras. Dessa forma, fazemos o Coombs direto, comprovamos diagnóstico e fazemos a transfusão com sangue ABO e Rh compatível.
- Ac Frios- IgM:
- Entre 0-10ºC, daí surgem as crioaglutinas
Se mostra bem mais raro, mas pode ocorrer um chamado "complexxo de ataque à membrana" (C5b-C9). Isso pode causar hemólise intravascular.
Causado pelo Antígeno I contra o grupo sanguíneo (I,i)
Diferente do anterior que ocorria principalmente pela ação dos receptores FcγRI e uma parte pelas células de Kupffer, nesse caso é só pelas células de Kupffer.- Clínica:
- A forma idiopática da doença é a famosa doença da crioglobulina
A causa mais comum identificada é a infecção pelo Mycoplasma pneumoniae
Para diagnóstico fazemos o coombs direto e a dosagem dos níveis séricos de crioglubulina
Tratamento: rituximabe
Clorambucil (refratário- imunossupressor ou agente alquilante)
- Anticorpo de Donath-Landsteiner:
- Anticorpo contra o antígeno P da membrana eritrocitária
É uma crio-hemolisina
Permite formação do complexo C5b-C9 que provoca alta entrada de água, e hemólise intravascular.
Quando se tem hemoglobinúria- chama-se síndrome da crio-hemoglobinúria paroxística
- Anemia Imuno-hemolítica farmacoinduzida:
- Tipo autoimune: alfametildopa e levodopa- a situação fisiopatológica é idêntica à do AHAI por IgG idiopática.
Tipo hapteno: quando uma molécula não proteica se liga a uma proteína do organismo e forma um complexo antigênico, é chamada de hapteno. Há dois subtipos de hemólise do tipo hapteno: 1) penicilina G cristalina quando mais de 10-20 milhões de U/d; 2) quinidina se liga à glicoproteína da membrana e acaba estimulando a IgM e ativando o complemento, causa hemólise intravascular (geralmente grave). Isso acontece também com as sulfas e a clorpromazina

Permissões neste sub-fórum
Não podes responder a tópicos